5q Spinal Muscular Atrophy (5q-SMA)
Spinal Muscular Atrophy, also known as SMA, affects 1/6000 to 1/10,000 live births and is a leading genetic cause of death in infant. It encompasses a group of inherited neuromuscular disorders which affect the nerve cells, called motor neurons, in the anterior horn cells of the spinal cord that control voluntary muscle movement such as crawling, walking, raising arms, holding objects with hands , moving head and neck around, breathing, and swallowing.
The most common form of SMA is caused by loss or mutation of the survival motor neuron 1 gene (SMN1) on chromosome 5. This predominated form is called 5q-SMA, which accounts for about 95% of SMA cases. The SMN1 gene is responsible for producing an important protein called survival motor neuron (SMN) protein, which are required for the proper functioning of motor neuron. As the SMN1 gene does not make enough SMN protein, the motor neurons break down, preventing the muscles from receiving proper signals from the brain. This in turn leads to progressive atrophy of the skeletal muscles.
There are other rare forms of SMA which are not caused by SMN1 gene mutation (such as SMA with Respiratory Distress, distal SMA, and etc.). These forms vary considerably in severity and are broadly called non 5q-SMA. Unless specify, the use of the word “SMA” in the content below refers to 5q-SMA only.
Types and symptoms
Each SMA children has different severity of weakness generally correlates with the age of onset. Weakness is greater proximally (muscles closer to the body such as shoulder, hips and upper back muscles) than distally (muscles not close to the body such as hands and feet) and generally greater in the legs than the arms. There are often loss of reflexes. In severe cases of SMA, the muscles responsible for breathing and swallowing may also be severely affected. SMA children have normal cognition and sensation.
SMA is commonly classified into 4 main clinical types (from Type I through Type IV) according to the age of onset and/or highest level of motor development milestone attained (Table 1). Milestones can include the ability to sit, stand, or walk. These “types” are not rigid categories, there is a wide spectrum of severity and considerable variability in the rate of progression within each type.
Table 1 – Clinical classification of SMA
| Type | Onset age | Life Span | Highest Motor Ability |
|---|---|---|---|
| Type I (severe) | Prenatal to first 6 months of infancy | < 2 yrs | Never sit independently |
| Type II (intermediate) | 6-18 months | >2 yrs | Sits alone; Never walk independently |
| Type III (mild) | 18 months -> 17 yrs old | Adult | Walk independently |
| Type IV (adult) | > 18 yrs old | Normal | Walk independently |
SMA type I, also known as the Werdnig-Hoffman disease or severe infantile SMA, is the most severe and common form accounting for 50-70% of childhood-onset SMA. Initial symptoms are often evidenced within the first 6 months of life (with some babies diagnosed even before birth) when the parents notice the baby fails to sit up, has weak cry or swallowing & feeding difficulties.
Infants with SMA type I are often described as “floppy babies” since their muscle tone is significantly reduced (hypotonia) manifesting a "frog-like" posture on lying. They never achieve independent sitting, roll over or crawl. They typically suffer from feeding and swallowing problems, and respiratory distress from recurrent aspiration causing severe pneumonia that could be an important cause of early death. Tongue fasciculation (the involuntary muscle twitching in the tongue) are also common. SMA type I progresses rapidly and death usually occurs before 2 years of age if the child does not have any ventilation support. However, with the timely use of supportive interventions, including application of non-invasive ventilation, mechanical insufflation-exsufflation cough assist device and the gastrostomy tube feeding, life expectancy has been markedly improved to late childhood and even adulthood. At the older age, children with SMA type 1 will also develop scoliosis (the curvature of the spine).
SMA type II, also known as Dubowitz diseases or intermediate SMA. Affected infants often develop symptoms between 6-18 months of age, although weak muscle tone may be noticed in the first few months after birth. Affected infants can sit without support (although some may lose this ability over time), but they are unable to walk independently. They also have decreased or absent deep tendon reflexes and distal hand tremors. Pulmonary and feeding problems, as well as scoliosis, are also common in type II patients but is less severe and have a more chronic and slowly progressive course than that seen in type I infants. The rate of progression among different type II patients can vary considerably. Life expectancy can range from early childhood to adulthood, depending on the severity of the patient's condition.
SMA type III, is also known as the Kugelberg-Welander disease. The affected patients develop symptoms between 18 months to 17 years of age. SMA type III children learn to walk independently, although approximately half of them will eventually loss their ambulation before adulthood. Most patients, over time, will eventually walk with increase waddling, have more frequent falls and more difficulty in getting up from a sitting position. They often have fine finger tremor. Feeding and swallowing problems during childhood are not common. Some of them will also develop scoliosis, requiring brace or surgical correction at later age. Life expectancy is not significantly different compared with normal population.
SMA type IV is characterized by adult onset. The majority of reported SMA type IV cases presenting symptoms after 30 years of age (although in some cases the diagnosis can be in their late teens and early adulthood) with normal mobility. Weakness is gradual, first affecting the thighs and hips before affecting the upper arms and the shoulders. Fatigue is common and patients often suffer cramps. Life expectancy is normal and respiratory and swallowing functions are rarely affected.
Genetic Cause
SMA is caused by the partial or complete deletion or small mutation of the Survival Motor Neuron 1 (SMN1) gene located on chromosome 5, which is responsible for the production of an important protein called Survival Motor Neuron (SMN) protein. SMN protein is found throughout the body but mostly in the spinal cord. The SMN protein plays an important role to maintain the normal function of the specialized motor nerve cells called motor neurons. These motor neurons are found in both the brainstem (part of the brain that is connected to the spinal cord) and the spinal cord and transmit signals from the brain and spinal cord to the skeletal muscles directing movement and muscle contraction. Without sufficient SMN protein, the motor neurons cannot be maintained in a healthy state and may even die, therefore attenuated the control of voluntary muscle movements which in turn results in weakness and wasting of the skeletal muscles. Muscles for speaking, swallowing, breathing, sitting and walking can all be affected.
The most common types of mutations are either lack of both SMN1 genes owing to partial or complete deletion or gene conversion of SMN1 that occur in approximately 95% of SMA patients. In gene conversion, the SMN1 gene is changed into a SMN2-like gene with the ‘C’ in exon 7 is changed into ‘T’. The remaining 5% of SMA patients have only one SMN1 gene that lack exon 7, but with the other SMN1 gene has a point mutation. (Figure 1a, b, c)
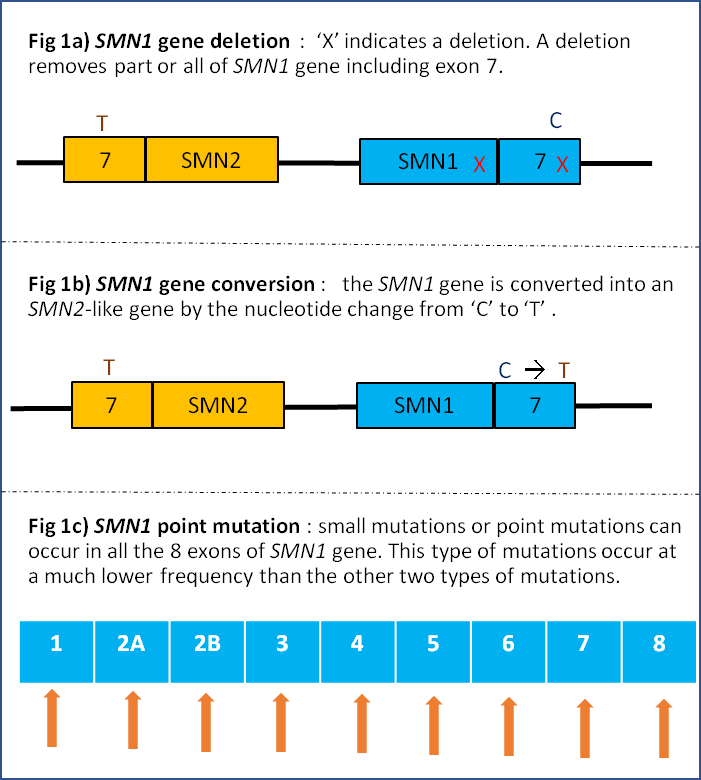
Another gene, known as the Survival Motor Neuron 2 (SMN2) gene, has a similar structure to SMN1 gene but with the nucleotide ‘C’ in exon 7 is changed into ‘T’, and only produces a small amount (about 10%) of fully functional SMN protein which can only partially compensate for the SMN protein deficiency caused by SMN1 gene mutation. The number of SMN2 copy varies in human but SMA patients always carry at least 1 copy of SMN2 gene.
Carrier
SMA has an estimated carrier frequency of 1 in every 40-60 people. It occurs through autosomal recessive inheritance, meaning that in general both parents are carriers (who have one healthy copy and one faulty copy of SMN1 gene) and the child inherits 2 faulty SMN1 genes, one from each parent, and develops SMA.
SMA carrier generally has no symptoms but the faulty gene can be passed on to their children. If both members of a couple are carriers, every single pregnancy has a 25% risk of giving birth to a SMA baby. Therefore, reproductive decision is sensitive, highlighting the need for genetic counselling on inheritance, recurrent risk and possible options for prenatal diagnosis of the future pregnancy.
Diagnosis
When diagnosis of 5q-SMA is suspected upon clinical examination, the first diagnostic test to be undertaken will be the SMN1 gene testing. Blood will be taken for the gene-targeted deletion/duplication study to decide on the dosage for SMN1 exon 7. If exon 7 is deleted from both SMN1 copies, the diagnosis of SMA is confirmed. If exon 7 is deleted from one copy of SMN1, whole sequence analysis will be performed on the other SMN1 gene to look for its point mutation, if any. If there is exon 7 deletion in one copy of SMN1 and point mutation in the other copy of SMN1, the diagnosis of SMA is again confirmed. SMN2 gene study checking the copy numbers will also be carried out.
On the other hand, if exon 7 are present in both SMN1 genes, one will need to consider other differential diagnoses requiring further neuromuscular diagnostic workup.
The pilot program of SMA newborn screening was started in Chinese University private genetic service in 2022. In October 2023, SMA newborn screening is also available to all public hospitals under the Hospital Authority. Currently, this program is extending to the private hospitals.
SMA newborn screening allows the earliest diagnosis of babies with SMA in the pre-symptomatic or early symptomatic stage. With the earlier starting of treatment, affected children can achieve a much better clinical outcome and even have a chance of living a normal life.
Table 2 below summarizes the aforementioned 3 possible outcomes of SMN1 gene-targeted deletion/duplication study.
Table 2. A child suspected to have SMA with SMN1 gene-targeted deletion/duplication study
| Number of full SMN1 gene | Conclusion / Further Diagnosis Testing Required |
|---|---|
0 |
With deletion or absence of both SMN1 genes, diagnosis of 5q-SMA is confirmed. |
| 1 | With absence of only 1 copy of SMN1 gene, one need to look for point mutation in the other SMN1 gene through whole gene sequencing If point mutation is present in one SMN1 gene, and there is deletion in the other SMN1 gene, the diagnosis of 5q-SMA is confirmed. If there is no point mutation in the remaining 1 copy of the SMN1 gene, one need to consider other differential diagnosis. |
| 2 or more | With the presence of 2 copies of SMN1 gene, 5q-SMA is unlikely. One need to consider other differential diagnoses and perform further testing such as creatine kinase (CK) level, electromyography (EMG), nerve conduction study (NCS) and , in certain cases, even muscle biopsy and multi-genes panel, to look for other underlying rare neuromuscular conditions with SMA-like presentation. |
Treatment
Spinraza (Nusinersen) is an antisense oligonucleotide that increase functional SMN protein level by targeting SMN2 gene. It is administered by intrathecal injection into the cerebrospinal fluid surrounding the spinal cord. In December 2015, Hong Kong participated in phase 3 clinical trial of Spinraza treatment for infant with SMA type 1 and 2 patients (SHINE and CHERISH studies). In December 2016, the US Food and Drug Administration (FDA) approved Spinraza as the first approved drug to treat SMA. Since April 2018, SMA type I patients in Hong Kong that are medically stable could receive nusinersen treatment under the Expanded Access Program (EAP). In September 2018, Spinraza is officially added to the HKSAR Department of Health registered list of pharmaceutical products. Due to its extreme high cost, the HKSAR government is providing the financial assistance through the Community Care Fund (CCF). Under the Hospital Authority, all eligible patients will be supported through the CCF Medical Assistance Programme, and the SMA expert panel will regularly review the progress of the patients under the SMA treatment program.
Evrysdi (Risdiplam) is an oral drug, also a small molecule, that enables the SMN2 gene to produce more functional SMN proteins. In August 2020, it was approved by the FDA to treat SMA patients. Since 2022, it is officially added to the HKSAR Department of Health registered list of pharmaceutical products and eligible patients are financially supported by CCF. The SMA expert panel will regularly review the progress of the patients receiving this treatment.
Zolgensma (Onasemnogene abeparvovec) is a one-time, intravenous gene therapy that deliver a functional SMN1 gene into patient’s motor neurons, enabling production of missing SMN protein. It was approved by FDA as the first gene therapy for SMA in May 2019. Zolgensma became available in Hong Kong since 2024 for patients with SMA diagnosed from newborn screening and those under 2 years of age. The SMA expert panel will regularly review the progress of those patients who have received this treatment.
Itvisma (onasemnogene abeparvovec-brve) is a one-time, intrathecal gene therapy for SMA. It has recently received FDA approval in November 2025, for treating adults and pediatric patients aged two years and older. It is not available in Hong Kong yet.
Investigatory drugs with different therapeutic strategies with ongoing clinical trials: These including Branaplam (RG7916), Tirasemtiv, SRK-015… Please check out SMA Europe for more information.

Standard of Care. Care for children with SMA is often best accomplished with the help of many specialists and primary care providers. Parents are key members of this team and are encouraged to participate as much as possible. The standard of care will also maximize the clinical effects of the SMA disease-modifying therapies.
SMA Care Management Family Guide SMA diseases severity vary with different types. Hence, care decisions shall be personalized based on the individual’s medical condition and functional ability. The family guide here contains detailed recommendations about the best standard of care on the following care areas (pulmonary care, feeding & nutrition, musculoskeletal care, mobility assessment and intervention). The guideline is prepared based on the Consensus Statement for Standard of Care in Spinal Muscular Atrophy drawn up by a large group of SMA experts that participated the International Conference on SMA Standard of Care (Wang CH et al. Journal of Child Neurology 2007;22(8):1027-49). An updated recommendation of standard of care has been recently published in the following two papers : Mercuri E et al. Neuromuscular Disorder 2018;28:103-115 |
Pulmonary care – SMA type I and type II children have weak intercostal muscles (muscles supporting the chest wall) leaving the diaphragm as the primarily breathing muscle. As a result, SMA type I and type II children suffer from chest wall and lung under development and are vulnerable to the following respiratory problems:-
- Impaired coughing so preventing clearance of mucus and other secretions in the airways which may lead to difficulty in breathing, easy development of pneumonia or even collapsed lung;
- Chronic hypoventilation (shallow and rapid breaths) during sleep;
- Aspiration pneumonia from recurrent silent aspiration of food or fluids due to the presence of feeding problem;
- Respiratory muscle weakness exacerbates by recurrent infections;
- Respiratory insufficiency due to weak respiratory muscles.
With respiratory problems being the leading causes of illness and the most common cause of death for SMA type I and type II patients, the respiratory specialty team will conduct regular pulmonary function assessment on the child and recommend the following interventions if the above mentioned respiratory problems occur :-
- Regular chest physiotherapy, postural drainage, use of cough assistance machine to mobilize and clear secretions or mucus;
- Regular breathing exercise to help strengthen the breathing muscles;
- Introduction of the non-invasive breathing device with BIPAP support to assist breathing;
- Introduction of invasive respiratory care (for severe cases or in medical emergency only) via an endotracheal tube that goes through the mouth (intubation) or directly into the trachea through a small incision in the neck (tracheotomy) to provide a secure airway to the lungs for efficient ventilation support;
- Completion of recommended immunization schedule including yearly seasonal flu vaccines to avoid preventable illness.
Gastrointestinal and Nutritional Care - SMA patients, especially SMA type I and II are susceptible to gastrointestinal and nutritional health issues caused by overall muscle weakness. As their jaw and oropharyngeal muscles are weak putting them at risk of incomplete chewing and easy inhalation of food and liquid into their airway instead of the esophagus. Children with SMA, especially for those of non-ambulatory status, often have difficulties moving food through the gastrointestinal tract and often have constipation requiring medical treatment. They may also develop problems such as acid reflux, vomiting and bloating, therefore vulnerable to aspiration.
Chewing and feeding problem can be initially managed with the use a semi-solid diet and thickened liquids to decrease the need of chewing. With the slower flow of the thickened liquid, the child can coordinate the swallowing better. An adequate dietary fiber with adequate fluid intake is also essential to lower the chance of constipation. They should also avoid high fat diet as high fat content in the food delay gastric emptying and increase the risk of gastroesophageal reflux. Occupational therapists and physiotherapists may advise on the best positioning and seating arrangement to facilitate feeding. If oral feeding and swallowing becomes unsafe or the oral food intake is always inadequate to meet the nutritional need, intervention of putting a feeding tube to provide the necessary nutrition and hydration will be necessary. This will include a temporary nasogastric tube (a tube that passes through the nose and the throat into the stomach) or a more permanent gastrostomy tube (placed surgically through the abdominal wall and into the stomach).
The growth and nutritional status of SMA children needs regular monitoring. SMA type I or severe SMA type II children without any feeding support are inclined to have under nutrition with poor weight gain due to their poor oral intake and the poor tolerance of feeds. On the other hand, SMA type I children on gastrostomy feeding, or mild SMA type II are at risk of over-nutrition, as they are taking in more than they need which will lead to overweight or even obesity. Nutrition management and care for children with SMA is complex and the dietary recommendation should be individualized and regular assessment by a dietitian is recommended.
Orthopedics Care and Rehabilitation - Since muscle weakness in the legs and arms may result in tightness of joints (a condition known as a contracture), regular stretching exercises, supported standing on stander, joint activity training, muscle mobility training, and use of joint braces, are helpful in preventing joint contractures. Rehabilitation equipment such as assistive walkers, manual or electrical wheelchairs are often needed in patients with non-ambulatory status or have limited walking ability with support.
Bone health Care and Intervention - Low bone mineral density and reduced bone volume are common in SMA patients. Monitoring of vitamin D level and DEXA scan evaluation for bone mineral density should be regularly performed. Bisphosphonate can be administered in patients with osteoporosis having low bone mineral density and history of bone fracture or vertebral collapse.
Hip subluxation and dislocation, and feet deformities are also common in SMA type I and type II patients. Scoliosis is also common in SMA type I and II, and can occurs in more severe SMA type III patients at a later age. An early use of spinal brace and a proper sitting posture with adequate head, neck and back support in the seating system, play an important role to prevent the rapid deterioration of the spinal curve and the pelvic obliquity. If the spinal curve becomes severe, a corrective spinal surgery may be needed.
Other Useful Information
Here is a list of additional websites that you may find more information on SMA:
- (廣州罕见病基因治療联盟 – China) 罕见病联盟官网
- (美儿SMA关爱中心 – China) 了解SMA - 美儿SMA关爱中心
- (脊髓肌肉萎縮症慈善基金 - HK) FSMA Hong Kong
- (香港肌健協會 - HK) 脊髓肌肉萎縮症(SMA)
- (香港神經肌肉疾病學會 - HK) 脊髓肌肉萎縮症
- (TREAT-NMD - UK) SMA Care Guide - TREAT-NMD
- (Cure SMA - US) Care Series Booklets for Spinal Muscular Atrophy (SMA)
- (Spinal Muscular Atrophy UK ) Homepage , Research & Treatment Overview
- (Muscular Dystrophy Association - US) Spinal Muscular Atrophy (SMA) - Diseases
- (Loop Community Muscular Dystrophy Association - AUS) Spinal Muscular Atrophy (SMA) - The Loop
- (TreatSMA - UK ) TreatSMA — UK Information & Community for Spinal Muscular Atrophy
- (SMA Europe) SMA Medicines Landscape , Clinical Trials for SMA